There are several types of DNA sequencing. Heritable and somatic DNA variants can be studied thanks to whole-genome sequencing (WGS), whole-exome sequencing (WES), and targeted sequencing. SNP and CGH arrays can be used to find genetic polymorphisms and copy-number variants, respectively, in addition to NGS data. Microbial community composition and functions can be analyzed using metagenomic whole-genome sequencing.
To answer research questions in both basic biology and biomedical contexts, we routinely analyze DNA sequence data. We have listed a few of the standard data analyses for DNA sequencing below. Send us a message, and we'll schedule a brief call with one of our experts for you if you're interested in learning how we can help you make the most of your DNA-seq data.
Variant analysis
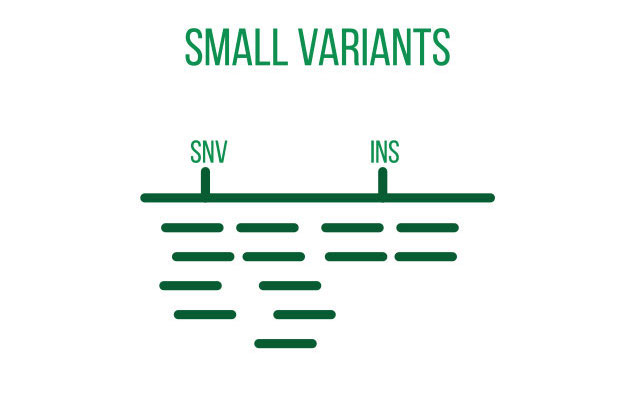
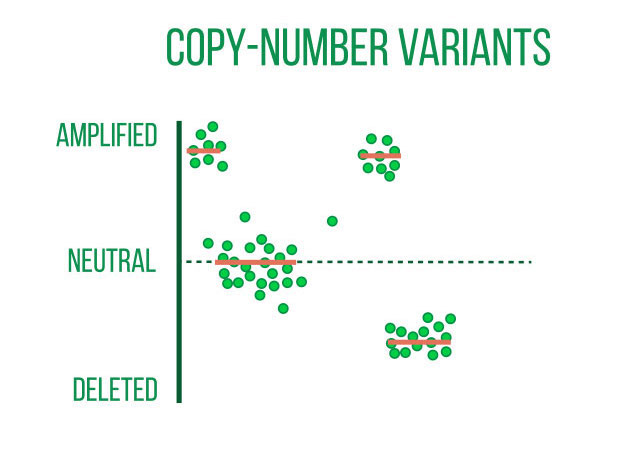
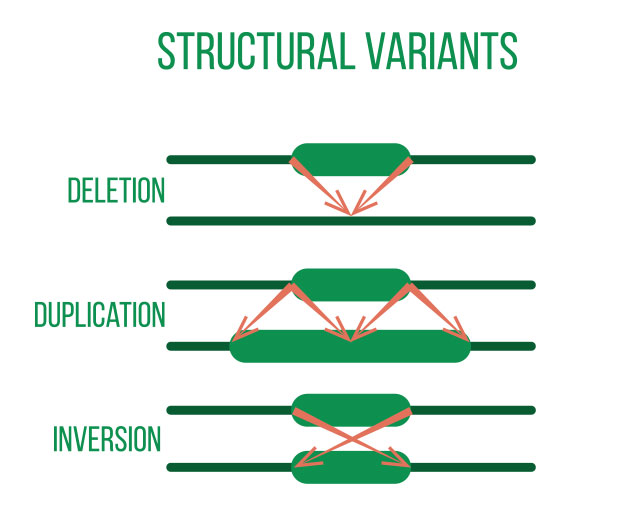
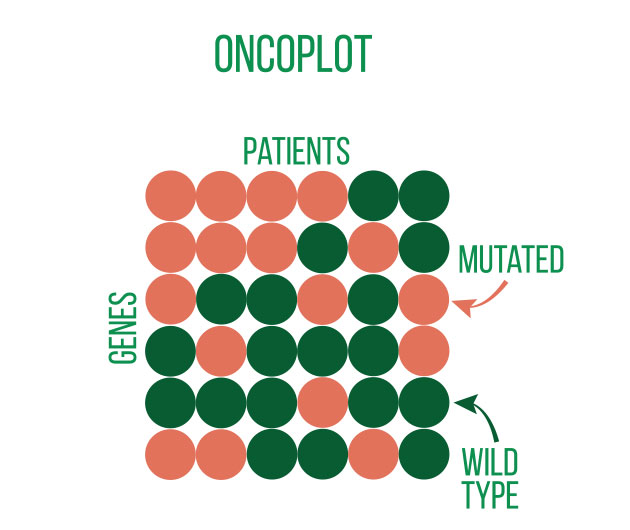
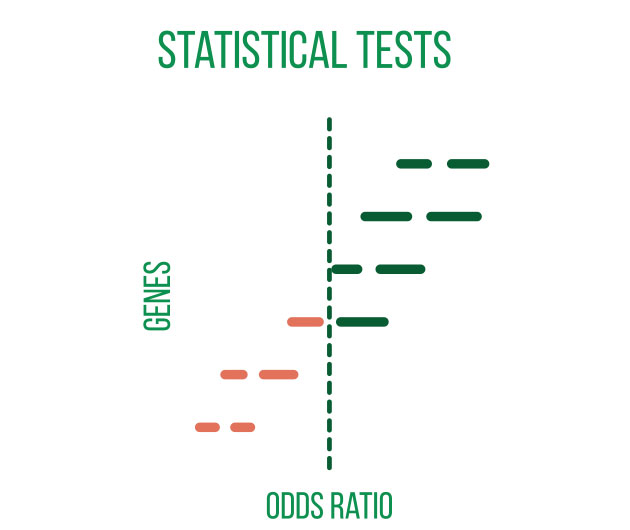
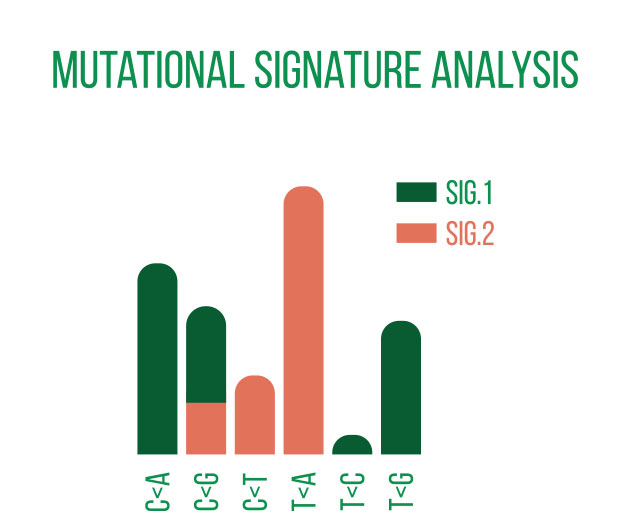
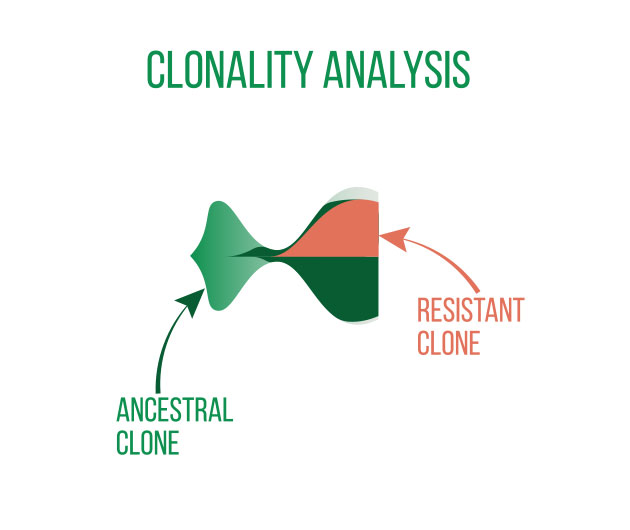
DNA sequencing is typically used to locate and examine genetic variations. Small nucleotide substitutions, insertions, deletions, copy-number changes, or structural variants are some examples of these variants. They could be somatic mutations or heritable polymorphisms.
It is customary to align the sequencing reads against a reference genome and perform quality control on the raw DNA-sequencing data before beginning a variant analysis. Then, using computational methods, variants that differ between the sample and a public reference or between different samples can be found.
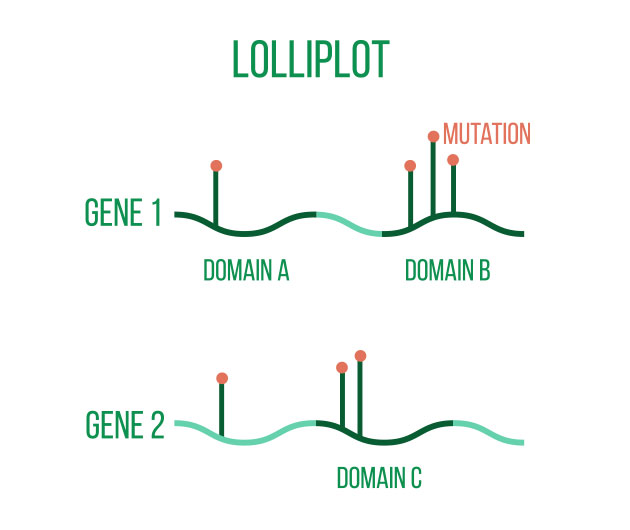
Annotating the discovered variants is an essential component of variant analysis. Annotations such as predicted effects on protein structure or gene regulation, allele frequencies (both in-sample and in public databases such as gnomAD), and predicted pathogeneicity allow for flexible selection or ranking of variants for subsequent analyses and interpretation.
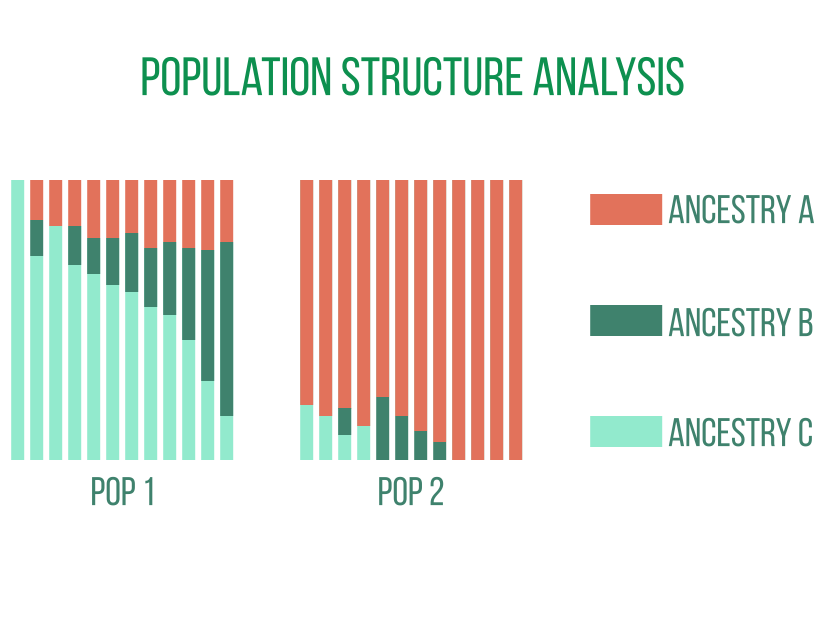

Population genetics
Genome-wide analyses of individuals drawn from related populations provide detailed insights into the structure, ancestry, and history of the populations. Genome assembly and annotation are frequent first steps in population genetic analyses of non-model organisms before locating genetic polymorphisms in the sampled populations. Studies of evolutionary phenomena like speciation and adaptation are aided by downstream analyses based on these polymorphisms and their allele frequencies.
Principal component analysis, genetic variation analysis within and between populations to identify loci affected by evolutionary selection, analyses of population admixtures, phylogenies, and demographic histories are examples of typical analyses.
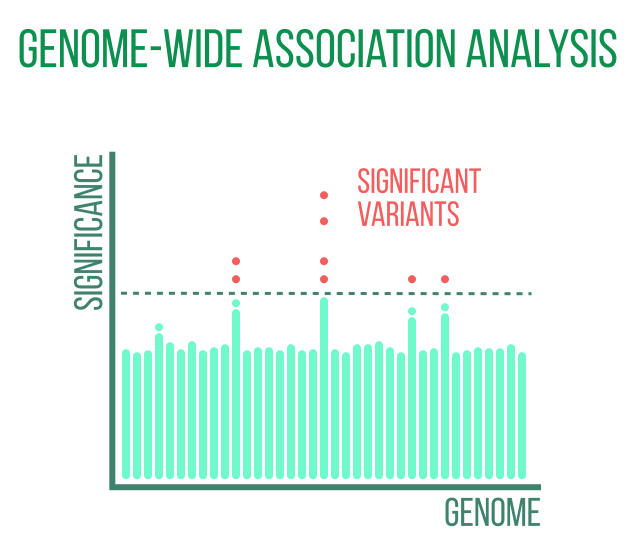
Genome-wide association analysis
Identification of genes and variants linked to pertinent phenotypes or diseases is the goal of biomedically motivated population-scale genetic analyses. The majority of diseases require large population-level sample sizes to achieve sufficient statistical power to identify associations, with the exception of a small number of diseases that are monogenic and strongly heritable. SNP-array or DNA-sequencing data from biobanks or other significant repositories are the foundation of these genome-wide association studies (GWAS).
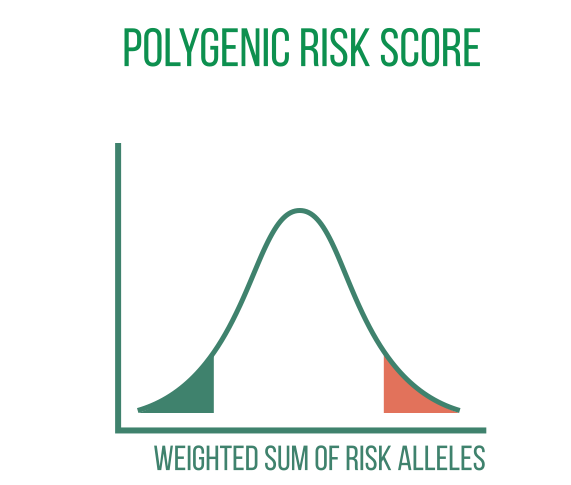
Summary statistics on the relationship between each individual variant and the investigated disease are produced by GWAS. Even when the disease is strongly heritable, individual variants in polygenic diseases may have very small effect sizes. In these situations, polygenic risk scores (PRS) can be used to add up the effects of numerous variants to produce a combined risk score that has clinical value.